





Genes 2025, 16(4), 439; https://doi.org/10.3390/genes16040439 (registering DOI) - 7 Apr 2025
Abstract
Background: Psychosis, particularly schizophrenia (SZ), is influenced by genetic and environmental factors. The neurodevelopmental hypothesis suggests that genetic factors affect neuronal circuit connectivity during perinatal periods, hence causing the onset of the diseases. In this study, we performed a genome-wide association study (GWAS)
[...] Read more.
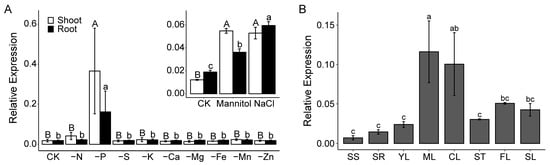
Background: Psychosis, particularly schizophrenia (SZ), is influenced by genetic and environmental factors. The neurodevelopmental hypothesis suggests that genetic factors affect neuronal circuit connectivity during perinatal periods, hence causing the onset of the diseases. In this study, we performed a genome-wide association study (GWAS) in a sample of the first episode of psychosis (FEP). Methods: A sample of 147 individuals diagnosed with non-affective psychosis and 102 controls were recruited and assessed. After venous blood and DNA extraction, the samples were genotyped. Genetic data underwent quality controls, genotype imputation, and a case-control genome-wide association study (GWAS). After the GWAS, results were investigated using an in silico functional mapping and annotation approach. Results: Our GWAS showed the association of 27 variants across 13 chromosomes at genome-wide significance (p < 1 × 10−7) and a total of 1976 candidate variants across 188 genes at suggestive significance (p < 1 × 10−5), mostly mapping in non-coding or intergenic regions. Gene-based tests reported the association of the SUFU (p = 4.8 × 10−7) and NCAN (p = 1.6 × 10−5) genes. Gene-sets enrichment analyses showed associations in the early stages of life, spanning from 12 to 24 post-conception weeks (p < 1.4 × 10−3) and in the late prenatal period (p = 1.4 × 10−3), in favor of the neurodevelopmental hypothesis. Moreover, several matches with the GWAS Catalog reported associations with strictly related traits, such as SZ, as well as with autism spectrum disorder, which shares some genetic overlap, and risk factors, such as neuroticism and alcohol dependence. Conclusions: The resulting genetic associations and the consequent functional analysis displayed common genetic liability between the non-affective psychosis, related traits, and risk factors. In sum, our investigation provided novel hints supporting the neurodevelopmental hypothesis in SZ and—in general—in non-affective psychoses.
Full article
(This article belongs to the Special Issue Genetics and Genomics of Psychiatric Disorders)
►
Show Figures

Figure 1













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}