


Hematol. Rep. 2025, 17(2), 18; https://doi.org/10.3390/hematolrep17020018 - 28 Mar 2025
Abstract
►
Show Figures
Leukemia is a heterogeneous group of hematologic malignancies characterized by distinct genetic and molecular abnormalities. Advancements in genomic technologies have significantly transformed the diagnosis, prognosis, and treatment strategies for leukemia. Among these, next-generation sequencing (NGS) has emerged as a powerful tool, enabling high-resolution
[...] Read more.
Leukemia is a heterogeneous group of hematologic malignancies characterized by distinct genetic and molecular abnormalities. Advancements in genomic technologies have significantly transformed the diagnosis, prognosis, and treatment strategies for leukemia. Among these, next-generation sequencing (NGS) has emerged as a powerful tool, enabling high-resolution genomic profiling that surpasses conventional diagnostic approaches. By providing comprehensive insights into genetic mutations, clonal evolution, and resistance mechanisms, NGS has revolutionized precision medicine in leukemia management. Despite its transformative potential, the clinical integration of NGS presents challenges, including data interpretation complexities, standardization issues, and cost considerations. However, continuous advancements in sequencing platforms and bioinformatics pipelines are enhancing the reliability and accessibility of NGS in routine clinical practice. The expanding role of NGS in leukemia is paving the way for improved risk stratification, targeted therapies, and real-time disease monitoring, ultimately leading to better patient outcomes. This review highlights the impact of NGS on leukemia research and clinical applications, discussing its advantages over traditional diagnostic techniques, key sequencing approaches, and emerging challenges. As precision oncology continues to evolve, NGS is expected to play an increasingly central role in the diagnosis and management of leukemia, driving innovations in personalized medicine and therapeutic interventions.
Full article
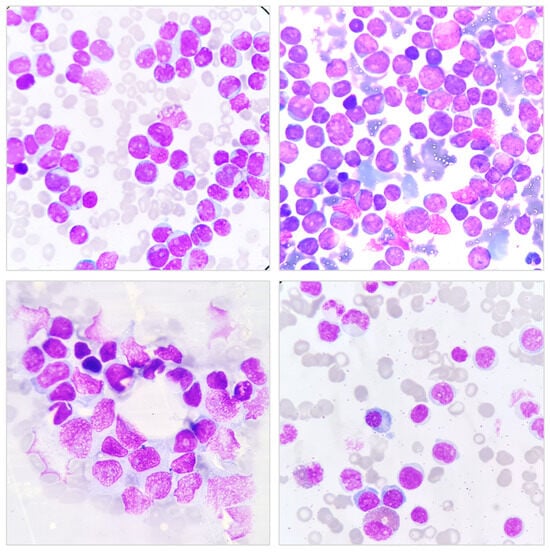
Figure 1













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}